APEC Study
Does the Gastric Acid Altering Effects of Proton Pump Inhibitors Effect Capecitabine Pharmacokinetics? (APEC Study)
Lead Investigator
Dr. Edmond Ang – Clinical Research Fellow, Auckland Cancer Trials Centre
APEC Study Details
Research Background
Capecitabine, is an oral prodrug of 5-FU. It is widely used in the management of gastrointestinal and breast cancer due to its efficacy and ease of administration. The absorption and bioavailability of capecitabine can be variable, as illustrated by the effects of food on its pharmacokinetic profile.1 Like other orally administered drugs, gastric pH may potentially alter the dissolution and absorption of capecitabine.
Proton Pump Inhibitors (PPIs) are the second most frequently prescribed medication in New Zealand.2 These drugs inhibit gastric acid secretion by blocking the proton pump of the gastric parietal cell and this remains a highly efficacious treatment for gastritis and gastro-oesophageal reflux disease. Concomitant use of PPI is common among patients with cancer (estimated between 20-55%) partly due to the fact that chemotherapy or concurrently administered supportive medications (e.g. steroids) can precipitate gastritis and reflux.3-4
Two recent publications on the management of gastrointestinal malignancies raised concerns about the potential interaction between Capecitabine and PPI. In a retrospective study of 298 patients on capecitabine monotherapy for colorectal cancer, five-year recurrent free survival (RFS) rates were inferior among patients on concomitant PPI therapy.5 In addition, a post-hoc analysis of the TRIO-013 trial (phase III randomised trial comparing capecitabine and oxaliplatin (CAPOX) with or without lapatinib in patients with HER2 positive metastatic gastroesophageal cancer) reported poorer progression free survival (PFS) and overall survival (OS) in patients on concomitant PPI use in the CAPOX alone arm.6
It is postulated that the pH altering effects of proton pump inhibitors could adversely alter the dissolution, absorption and hence bioavailability of capecitabine, leading to diminished treatment efficacy. This is in part supported by invitro experiments conducted by Roche which showed that capecitabine tablets take longer to dissolve and disintegrate in basic solution.1 However, there is paucity of good quality pharmacokinetic studies assessing this potentially important interaction.
A recent Japanese study that prospectively examined the effects of rabeprazole on capecitabine and its metabolites failed to demonstrate any potentially significant pharmacokinetic impact.7 The study design was however flawed with inadequate control and a failure to take into account the differences in baseline characteristics (e.g. renal function) and dosage of capecitabine between the two study groups. In addition, alternative modes of potential drug-drug interaction (e.g. effects of PPI on capecitabine metabolite transfer into cells) have not been appropriately investigated.
Due to the importance and the potential impact of this clinical question on cancer treatment safety and efficacy in New Zealand, The Medicines Adverse Reactions Committee of New Zealand reviewed this potential drug interaction in December 2019. In an ensuing report, the committee cautioned that such an interaction is possible but fell short of making any firm recommendation, admitting that the clinical significance is yet to be known.
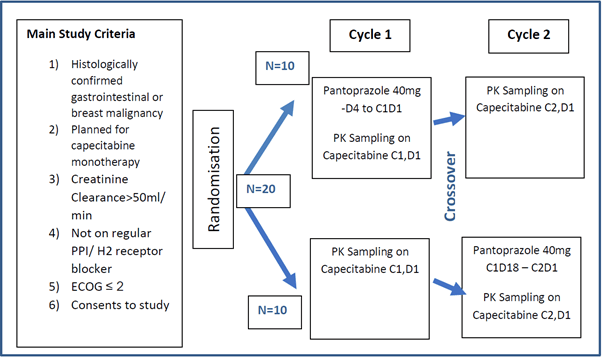
APEC Study Schema: Prospective, randomised, cross-over study to assess the pharmacokinetic effects of pantoprazole on capecitabine.
Expected Recruitment Numbers
Twenty patients receiving capecitabine monotherapy for the treatment of histologically confirmed gastrointestinal or breast cancer. Historically, similar pharmacokinetic (PK) studies have used cohort sizes of between 10-20 subjects. Given the cross over design of this study, a target recruitment of 20 is deemed adequate as each subject will serve as their own control.
Resource Available
This project will be conducted by the cancer and blood research team at the Auckland Cancer Trials Centre, a dedicated standalone early phase clinical trials facility located within Auckland City Hospital. The team comprises of Dr Sanjeev Deva (consultant medical oncologist with a special interest in gastrointestinal cancer and early phase/ translational research), clinical research fellows, research nurses and supportive staff.
The study will be conducted in mutual collaboration with Associate Professor Nuala Helsby, a leading scientist and researcher in chemotherapeutic drugs. Associate Professor Helsby and her research laboratory team at the Department of Molecular Medicine and Pathology University of Auckland will provide the scientific and technical expertise for the study, including processing and analysing pharmacokinetic blood samples and data analysis.
The cancer and blood services at Auckland City Hospital remains the sole public provider of cancer care to Auckland’s population of 1.6 million. Approximately 250 patients are placed on capecitabine monotherapy at Auckland City Hospital yearly. Due to the favourable volume of patients at our facility, study enrolment is anticipated to be completed within a year.
Methodology
Twenty patients receiving capecitabine monotherapy for the treatment of histologically confirmed gastrointestinal or breast cancer will undergo pharmacokinetic blood sampling during day 1 of their first two cycles of capecitabine. Consented study participants will be randomised to have pantoprazole either before their first or second cycle of capecitabine. Historically, similar pharmacokinetic (PK) studies have used cohort sizes of between 10-20 subjects. Given the cross over design of this study, a target recruitment of 20 is deemed adequate as each subject will serve as their own control.
A minimum of 3 days dosing is required to reach steady state inhibition of gastric acid secretion by PPIs.9 Pantoprazole will be dosed at 8am daily starting a minimum of 4 days prior and continued to day 1 of either cycle 1 or 2 of capecitabine, depending on subject randomization. Capecitabine dosing and pharmacokinetic blood sampling will occur on day 1 of each cycle after which pantoprazole will be stopped on day 2, of either cycle. The steps above are to ensure that subjects are exposed to a minimum duration of pantoprazole in case of any potential impact of pantoprazole on capecitabine efficacy while ensuring test validity through adequate pantoprazole action at the time of PK sampling.
Since the half-life of pantoprazole is short (0.8- 2 hours) and the half-life of the parietal cell proton pump (covalently inhibited by PPI) is 54 hours, the effects of previous pantoprazole exposure on gastric acid inhibition will have ‘washed out’ in the group randomised to receive pantoprazole prior to cycle 1.9 To minimize the known effects of food on variable capecitabine absorption, a standardised breakfast will be provided on the first day of cycle 1 and 2 for each individual subject. Capecitabine will be administered 15 minutes after completion of breakfast.
This project will be conducted at the Auckland Cancer Trials Centre, Auckland City Hospital in mutual collaboration with Associate Professor Nuala Helsby, a leading scientist and
researcher in chemotherapeutic drugs. Associate Professor Helsby and her research laboratory team at the Department of Molecular Medicine and Pathology, University of Auckland will provide the scientific and technical expertise for the study.
Pharmacokinetic blood samples will be drawn into 8mL EDTA tubes at the Auckland Cancer Trials Centre. To avoid conversion of 5’DFCR into 5’DFUR, blood samples are immediately spiked with a cytidine deaminase inhibitor (THU). The sample is then centrifuged and the resulting plasma transferred into fresh tubes prior to storage at -80oC. The plasma samples will be transferred to the University of Auckland. The plasma samples will be analysed by LC/MS and the concentrations of capecitabine and its metabolites recorded.
Bioanalysis of the pharmacokinetic data will be performed by Associate Professor Helsby and her team, using a validated published method for bioanalysis of capecitabine and metabolites. To take to account inter-individual variability, pharmacokinetic parameters will be normalised to the dosage of capecitabine received. The pharmacokinetic parameters (Tmax, Cmax, AUC0-last time; AUC0-∞) of capecitabine and its metabolites with and without PPI will be tabulated for each patient. To determine the effect of concomitant PPI on capecitabine PK, each patient will act as their own control and the PK parameters will be reported as ratio of no-PPI: plus-PPI. Descriptive statistics (median, 95%CI) of these ratios will be assessed using the Wilcoxon matched pairs signed rank test, p values > 0.05 will be considered statistically significant. This non-parametric analysis will be used, due to the well-established non-normal variation in capecitabine PK disposition.
The cancer and blood services at Auckland City Hospital remains the sole public provider of cancer care to Auckland’s population of 1.6 million. Approximately 250 patients are placed on capecitabine monotherapy at Auckland City Hospital yearly. Due to this favourable volume of patients at our facility, study enrolment is anticipated to be completed within a year.
Inclusion/ Exclusion Criteria
Inclusion:- 18 years and older
- Must be able to understand and sign consent.
- ECOG functional status score of equal or <2
- Willing to accept intense PK sampling on D1 cycle 1 and 2 of capecitabine
- Willing to have standardized breakfast on D1 cycle 1 and 2 of capecitabine
- Willing to receive Pantoprazole
- Contraindications or intolerance to PPI.
- Proton pump inhibitor use within 7 days of capecitabine dosing.
- Antacid or H2 receptor blocker use 14 days prior to capecitabine dosing.
- Significant organ dysfunction precluding capecitabine therapy
- Neutrophil>1.5, Hb>9g, Platelet>75000
- Bilirubin<1.5x ULN, ALT, AST <2.0 ULN,
- Creatinine clearance calculated by Cockcroft Gault >50ml/min
- Concomitant use of drugs known to be strong inducers/inhibitors of CYP2C19
- Known dihydropyrimidine dehydrogenase (DPD) deficiency or any Grade 3 toxicities from previous exposure to capecitabine.
- Serious concurrent medical condition that is deemed to influence ability to safely participate in study
- Significant gastritis/ gastro-oesophageal reflux necessitating treatment
- Previous gastric surgery
- Unable to fast from midnight before standardised breakfast prior to PK studies on day 1 cycle 1 and 2.
Research Aims
Reasons for doing the study
New Zealand has one of the highest rates of colorectal cancer in the world. Capecitabine is used as an oral replacement for infusional 5-Flurouracil in both adjuvant and palliative chemotherapy for colorectal cancer. Proton Pump Inhibitors (PPIs) are the second most frequently prescribed medication in New Zealand. Concomitant use of PPI is common among patients with cancer (estimated between 20-55%) partly due to the fact that chemotherapy or concurrently administered supportive medications (e.g. steroids) can precipitate gastritis and reflux.
Two recent publications raised concerns about the potential interaction between Capecitabine and PPI. It is postulated that the pH altering effects of proton pump inhibitors could adversely alter the dissolution, absorption and hence bioavailability of capecitabine, leading to diminished treatment efficacy and poorer outcomes. To date, there is paucity of good quality PK studies assessing this potential interaction.
This study seeks to answer a concern around day to day oncology practice that may have significant implications for thousands of patients with gastrointestinal cancer across New Zealand and around the world. Such is the importance and the urgency to clarify these concerns that The Medicines Adverse Effect Committee specifically reviewed this potential interaction in December 2019, falling short of issuing a firm recommendation given the lack of rigorous research to accept or refute these concerns. The research team is of the impression that the more frequent use of capecitabine in New Zealand (as compared to other health jurisdictions such as the United States) makes it important for us to address the question locally. Moreover, besides a single Japanese study with significant methodological flaws, we are unaware of any other pharmacokinetic studies addressing the same question.
In the short term, this study will contribute to a pool of scientific evidence that would either support or refute the concerns raised about the effects of PPI on the pharmacokinetics of capecitabine. This may also provide direction for future research into the question. In the long term, this study, alongside other related studies, will hopefully provide definitive answers on whether to permit or discourage the concurrent use of proton pump inhibitors and capecitabine.
Objective
The primary objective of this study is to determine if the concomitant use of Pantoprazole changes the pharmacokinetic profile (Cmax, Tmax, and AUC) of capecitabine and its metabolites (5’DFCR, 5”DFUR, 5FU and FBAL).